Biological Foundations for Bioinformatics
The Central Dogma of Molecular Biology
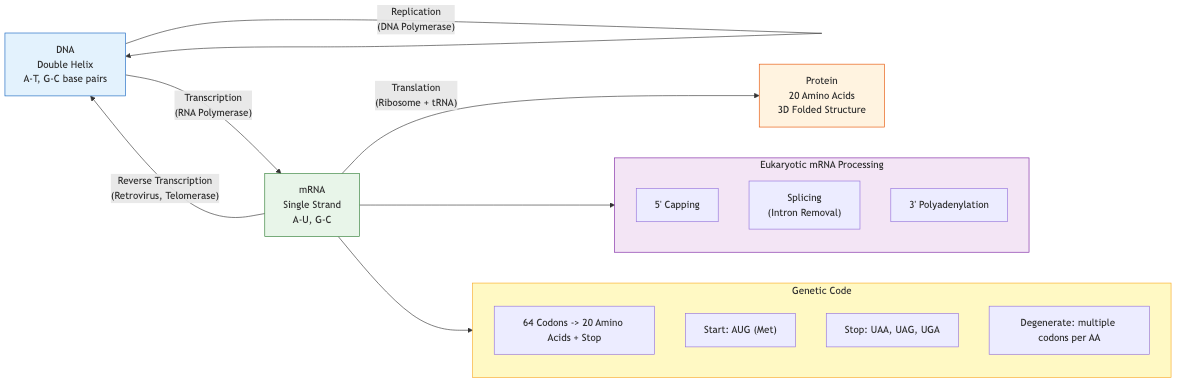
The central dogma describes information flow in biological systems: DNA is transcribed into RNA, which is translated into protein. This unidirectional flow (with known exceptions like reverse transcription) underpins all of computational biology.
DNA Structure and Function
DNA is a double-stranded helical polymer of deoxyribonucleotides. Each nucleotide consists of a deoxyribose sugar, a phosphate group, and one of four nitrogenous bases: adenine (A), thymine (T), guanine (G), or cytosine (C). Watson-Crick base pairing (A-T with two hydrogen bonds, G-C with three) creates the antiparallel double helix. The 5' to 3' directionality of each strand is critical for replication and transcription.
- Replication: Semi-conservative, catalyzed by DNA polymerase III (prokaryotes) or polymerases alpha/delta/epsilon (eukaryotes)
- Supercoiling: Managed by topoisomerases; relevant to genome topology and chromatin organization
- Telomeres: Repetitive sequences (TTAGGG in humans) protecting chromosome ends, maintained by telomerase
RNA Biology
RNA uses ribose and substitutes uracil (U) for thymine. Key RNA species:
- mRNA: Carries coding information; undergoes 5' capping, polyadenylation, and splicing in eukaryotes
- tRNA: Adapter molecules with anticodon loops; aminoacyl-tRNA synthetases charge them with cognate amino acids
- rRNA: Structural and catalytic component of ribosomes (16S/18S for small subunit, 23S/28S for large)
- Non-coding RNA: miRNA (~22 nt, post-transcriptional silencing), lncRNA (>200 nt, diverse regulatory roles), siRNA, piRNA, snoRNA, circRNA
Proteins
Proteins are polymers of 20 standard amino acids linked by peptide bonds. Each amino acid has an amino group, carboxyl group, and a variable R-group determining its physicochemical properties (hydrophobic, polar, charged, aromatic). Protein function depends on three-dimensional structure, which is determined by primary sequence through thermodynamic folding.
The Genetic Code
The genetic code maps 64 codons (triplets of nucleotides) to 20 amino acids plus three stop signals. Key properties:
| Property | Description |
|---|---|
| Degeneracy | Most amino acids encoded by multiple codons (wobble position) |
| Universality | Nearly universal across life; exceptions in mitochondria, Mycoplasma, ciliates |
| Non-overlapping | Codons read sequentially without overlap in standard translation |
| Unambiguous | Each codon specifies exactly one amino acid |
Codon usage bias: Organisms preferentially use certain synonymous codons, correlated with tRNA abundance. The Codon Adaptation Index (CAI) quantifies this bias and predicts expression levels.
Gene Expression and Regulation
Transcription
Prokaryotic transcription uses a single RNA polymerase with sigma factors for promoter recognition (-10 and -35 elements). Eukaryotic transcription employs three polymerases (Pol I: rRNA, Pol II: mRNA, Pol III: tRNA/5S rRNA) with complex promoter architecture (TATA box, CpG islands, enhancers, silencers).
Post-Transcriptional Processing
- Splicing: Intron removal by the spliceosome (U1, U2, U4, U5, U6 snRNPs); alternative splicing generates proteome diversity (>95% of multi-exon human genes)
- RNA editing: Adenosine-to-inosine (ADAR) and cytidine-to-uridine deamination
Translation
Ribosomes decode mRNA in three phases: initiation (Shine-Dalgarno/Kozak sequence recognition), elongation (codon-anticodon matching, peptide bond formation), termination (release factors at stop codons). Post-translational modifications (phosphorylation, glycosylation, ubiquitination) expand functional diversity.
Genome Organization
Prokaryotic Genomes
Typically single circular chromosomes (1-10 Mb), gene-dense (~85-95% coding), organized into operons with polycistronic mRNA. Horizontal gene transfer via conjugation, transduction, and transformation drives genomic plasticity. Plasmids carry accessory genes (antibiotic resistance, virulence factors).
Eukaryotic Genomes
Linear chromosomes with centromeres and telomeres. The human genome (~3.2 Gb) is only ~1.5% protein-coding. Major non-coding components:
- Repetitive elements: SINEs (Alu, ~11%), LINEs (L1, ~17%), DNA transposons (~3%), LTR retrotransposons (~8%)
- Regulatory sequences: Enhancers, silencers, insulators, locus control regions
- Structural elements: Centromeric satellite DNA, heterochromatin
Chromatin and Epigenetics
DNA wraps around histone octamers (H2A, H2B, H3, H4) forming nucleosomes. Epigenetic modifications regulate gene expression without altering DNA sequence:
- DNA methylation: CpG dinucleotides; 5-methylcytosine associated with silencing; maintained by DNMT1, established by DNMT3a/b
- Histone modifications: Acetylation (activation, HATs/HDACs), methylation (context-dependent, e.g., H3K4me3 = active, H3K27me3 = repressive), phosphorylation, ubiquitination
- Chromatin remodeling: ATP-dependent complexes (SWI/SNF, ISWI, CHD, INO80) reposition nucleosomes
Mutations and Variation
Mutation Types
- Point mutations: Transitions (purine-purine or pyrimidine-pyrimidine), transversions (purine-pyrimidine)
- Insertions/deletions (indels): Frameshifts if not multiples of 3 in coding regions
- Structural variants: Inversions, translocations, duplications, copy number variations (CNVs)
- Microsatellite instability: Expansion/contraction of short tandem repeats (STRs)
Functional Impact
Variants are classified by consequence: synonymous, missense (conservative vs. radical), nonsense, splice-site, regulatory. Pathogenicity prediction tools (SIFT, PolyPhen-2, CADD, REVEL) integrate evolutionary conservation and structural features.
Biological Databases
Primary Sequence Databases
- GenBank/ENA/DDBJ: INSDC partnership; nucleotide sequences with annotations; >2 billion sequences
- UniProt: Protein sequences; UniProtKB/Swiss-Prot (curated, ~570K entries) and TrEMBL (automated, >200M entries)
- RefSeq: NCBI reference sequences with non-redundant, curated records
Structure Databases
- PDB (Protein Data Bank): 3D macromolecular structures from X-ray crystallography, cryo-EM, NMR; >200K structures
- AlphaFold DB: Predicted structures for >200M proteins
- SCOP/CATH: Hierarchical protein structure classification
Functional and Pathway Databases
- Gene Ontology (GO): Controlled vocabulary for molecular function, biological process, cellular component
- KEGG: Metabolic pathways, signaling networks, disease associations
- Pfam/InterPro: Protein domain families and functional annotations
- OMIM: Mendelian disease-gene relationships
Bioinformatics File Formats
Sequence Formats
FASTA: Header line starting with > followed by identifier, then sequence on subsequent lines. Simple, universal, no quality information.
>sp|P00533|EGFR_HUMAN Epidermal growth factor receptor
MRPSGTAGAALLALLAALCPASRALEEKKVCQGTSNKLTQLGTFEDHFLSLQRMFNNCEVV
FASTQ: Extends FASTA with per-base quality scores (Phred+33 encoding). Four lines per read: header (@), sequence, separator (+), quality string. Quality score Q = -10 log10(P_error); Q30 means 1 in 1000 error probability.
Alignment Formats
SAM/BAM: Sequence Alignment/Map format. SAM is tab-delimited text; BAM is the binary compressed equivalent. Fields include QNAME, FLAG (bitwise: mapped, paired, strand), RNAME, POS, MAPQ, CIGAR string (alignment operations: M/I/D/S/H/N), mate information, sequence, quality. The CIGAR string encodes the alignment compactly (e.g., 50M2I30M1D20M).
CRAM: Reference-based compression of alignment data; more space-efficient than BAM.
Variant Formats
VCF (Variant Call Format): Stores genomic variants. Header lines (##) specify metadata, INFO/FORMAT field definitions, and reference genome. Data lines contain CHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO, and per-sample genotype fields. Multi-allelic sites, structural variants, and phasing information are supported.
BED: Tab-delimited genomic intervals (chrom, start, end) with optional name, score, strand fields. Zero-based, half-open coordinate system.
Annotation Formats
GFF3/GTF: Gene annotations with nine columns (seqid, source, type, start, end, score, strand, phase, attributes). GTF uses a structured attribute format; GFF3 uses key=value pairs with hierarchical parent-child relationships.
Computational Considerations
Biological data exhibits particular computational properties: sequences are strings over small alphabets (4 for DNA, 20 for protein), genome-scale analyses involve gigabytes to terabytes of data, and biological signal is often subtle against a background of evolutionary noise. Understanding these biological foundations is essential for correct algorithmic design and meaningful interpretation of computational results.